PyData入門¶
Pythonでデータ解析をする時の、全体的な話です。今後適宜、資料を改変していきます。 SPARQLthonでの発表 での発表がもとになっています。
準備¶
以下のモジュールを使えるようにしておきます。 これから環境を構築するのであれば、これらすべてを一度にインストールできる、Anaconda の利用が便利です。
-
Pythonインタラクティブシェルの機能強化版。コードと結果を1つにまとめて保存できるので便利。
-
グラフ描画のためのライブラリ
-
表やベクトル形式のデータを扱うための、多彩で高性能な機能が満載のライブラリ
-
機械学習アルゴリズムを手軽に使えるライブラリ
Pandasを使ってみる¶
データの読み込みと整形¶
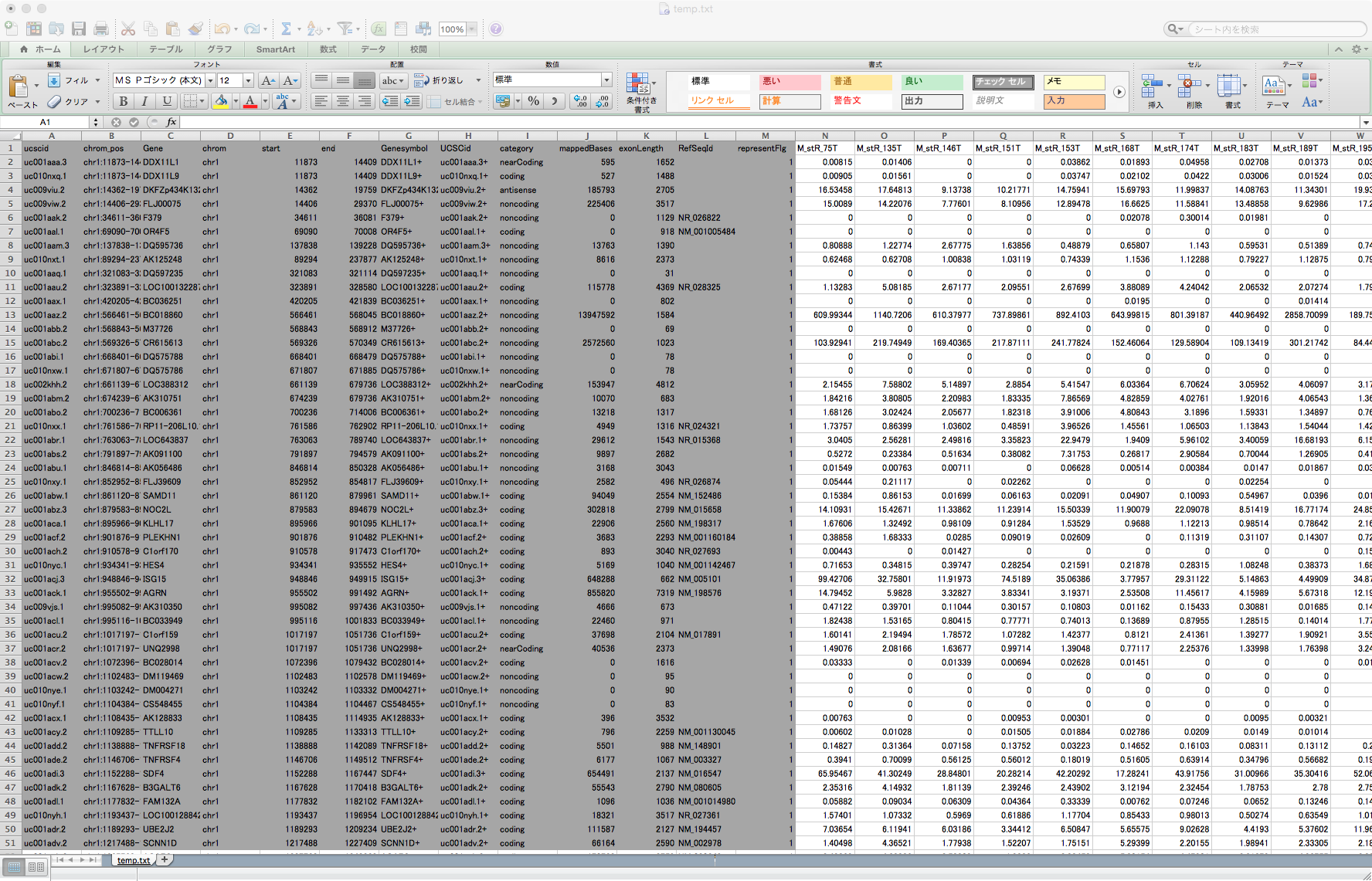
CSVデータをエクセルで表示
このデータは、肝臓ガンの患者さんの遺伝子発現データです。行方向に遺伝子が並んでいます。列には、患者さんが並んでいますが、背景が灰色になっている列は、患者さんに関係なく、遺伝子の付加的な情報です。タブ区切りのテキストデータになっているこのファイルを、pnadasで読み込むには、次のようにします。
import pandas as pd
_exp_data = pd.read_csv('pivot.txt',sep='\t',index_col=2)
ファイル名の後に、区切り文字を指定します。index_colで、読み込んだあと、行の名前になる列を指定できます。関数の戻り値は、pandas.DataFrame型で、ちょうどエクセルの1つのシートと同じような表形式のデータになります。
_exp_data
IPython notebookでは、これだけで簡単に中身を確認することができます。
列名を取得する。
_exp_data.columns
Index([u'ucscid', u'chrom_pos', u'chrom', u'start', u'end', u'Genesymbol',
u'UCSCid', u'category', u'mappedBases', u'exonLength',
...
u'M_stR_538N', u'M_stR_548N', u'M_stR_553N', u'M_stR_576N',
u'M_stR_581N', u'M_stR_612N', u'M_stR_627N', u'M_E_54N_DR',
u'M_E_712N_DR', u'M_E_742N_DR'],
dtype='object', length=203)
行の名前を取得する。
_exp_data.index
Index([u'DDX11L1', u'DDX11L9', u'DKFZp434K1323', u'FLJ00075', u'F379',
u'OR4F5', u'DQ595736', u'AK125248', u'DQ597235', u'LOC100132287',
...
u'XLOC_014495', u'XLOC_014497', u'XLOC_014499', u'XLOC_014500',
u'XLOC_014502', u'XLOC_014507', u'LINC00281', u'XLOC_014503',
u'XLOC_014504', u'XLOC_014505'],
dtype='object', name=u'Gene', length=38368)
どちらも、pandasのIndex型です。
特定の列にアクセスする。
_exp_data['chrom']
または
_exp_data.chrom
行を指定するときは、ix
_exp_data.ix['DDX11L1']
列名で全体をソートすることも可能。
_exp_data.sort('chrom')
列名のリストを渡せば、複数の列でソートすることも可能。
_exp_data.sort(['chrom','Genesymbol'])
数字だけのデータにしたいので、何列目からデータが入っているのか確認。
for i,v in enumerate(_exp_data.columns):
print('{}\t{}'.format(i,v))
# Rでもお馴染みのスライス
sample_ids = _exp_data.columns[12:]
12列目からの列名をsample_idsに格納。
# 列名で絞り込んで新たなDataFrameをexp_dataと命名
exp_data = _exp_data[sample_ids]
列の名前が、M_stR_75TやM_stR_135Tなどとなっていますが、最後の1文字がTの時は肝臓がんのデータ、Nのときは正常細胞のデータなので、この列名を、(75,T)や(135,T)などのタプルに変換。データ特有の変換なので、あまり気にしなくてもOKです。
def rename(c, _info, maps):
for v in maps:
if c in _info[v[0]].values:
return (_info[(_info[v[0]] == c)].index[0],v[1])
raise ValueError('{}は指定の列の範囲に見付かりません。'.format(c))
from functools import partial
exp_rename = partial(rename, _info=info, maps=[('dRNA_T','T'),('dRNA_N','N')])
exp_data.columns = list(map(exp_rename, exp_data.columns))
このように、列名や行名は、上書きできます。
ちょっとした統計処理¶
# サンプル(列)ごとの平均
exp_data.mean()
(75, T) 11.351534
(135, T) 12.739927
(146, T) 11.572280
(151, T) 12.490965
(153, T) 12.663369
...
# 遺伝子(行)ごとの平均
exp_data.mean(1)
Gene
DDX11L1 0.040685
DDX11L9 0.040093
DKFZp434K1323 11.384697
FLJ00075 10.763521
OR4F5 0.012345
...
引き数を省略すると0の意味。行列なので、引き数が0の時は、行方向に計算が進む。なので、最終的に列の平均が得られる。列方向に計算が進めば、行の平均が得られるという訳です。他の問う計量も計算できます。stdで標準偏差など。
#グラフを描画するための準備
%matplotlib inline
import pylab
# 遺伝子ごとの平均のヒストグラム
pylab.hist(exp_data.mean(1))
(array([ 3.83460000e+04, 1.30000000e+01, 2.00000000e+00,
1.00000000e+00, 4.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
2.00000000e+00]),
array([ 0. , 2580.05383807, 5160.10767614, 7740.1615142 ,
10320.21535227, 12900.26919034, 15480.32302841, 18060.37686648,
20640.43070454, 23220.48454261, 25800.53838068]),

# 出力を変数で受け取って
# binを細かく
_val = pylab.hist(exp_data.mean(1),bins=50)
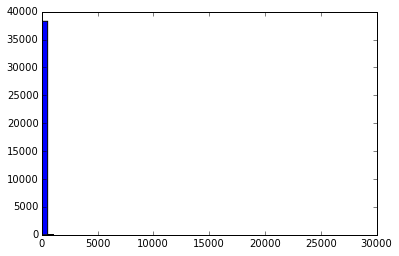
# 平均がすごいデカイ奴がいる。
# 誰?
exp_data.mean(1) > 5000
Gene
DDX11L1 False
DDX11L9 False
DKFZp434K1323 False
FLJ00075 False
F379 False
# 条件にあう行(遺伝子)だけが抽出できる
exp_data[exp_data.mean(1) > 5000]
#すべてのデータをlogに変換
exp_log = exp_data.applymap(pylab.log10)
# 自分用の関数を作る
my_log = lambda x:pylab.log10(x) if x > 1e-6 else pylab.log10(1e-6)
exp_log = exp_data.applymap(my_log)
0がマイナス無限大になってしまうので、適当なところで、打ち止めするために関数を自作しています。
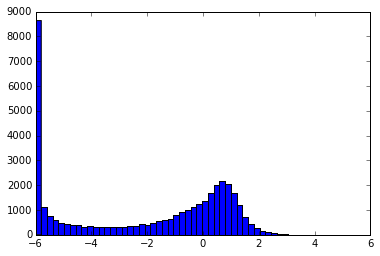
# 遺伝子ごとの平均のヒストグラム
_val = pylab.hist(exp_log.mean(1),bins=50)
#行の平均が-4より大きいものだけを残す
exp_log_m4 = exp_log[exp_log.mean(1)>-4]
_val = pylab.hist(exp_log_m4.mean(1),bins=50)
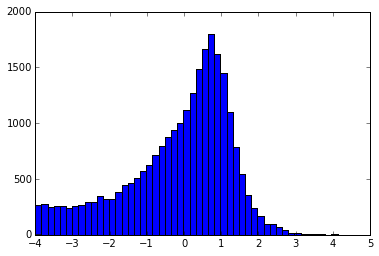
ヒストグラムからデータがフィルタリングされているのが分かります。
# 数が多いとデモデータとしては面倒なので、800行に削減
import random
idx = random.sample(exp_data_m4_s05.index,800)
data = exp_data_m4_s05.ix[idx]
データの可視化¶
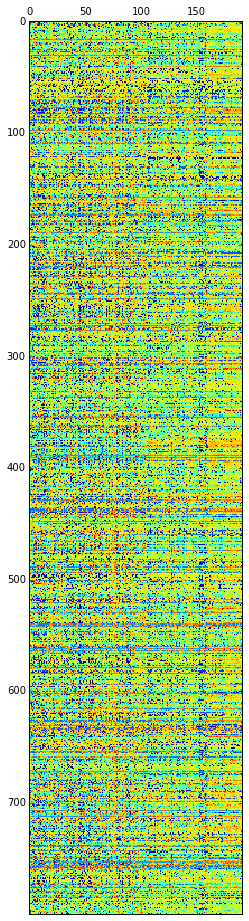
# ヒートマップ表示
pylab.matshow(data)

# データの標準化
# 遺伝子ごとに、平均値を引いて標準偏差で割る
_mean = data.mean(1)
_std = data.std(1)
ndata = data.subtract(_mean,0).div(_std,0)
# 色付けの範囲をvmin、vmaxで指定できる
pylab.matshow(ndata,vmin=-2,vmax=2)
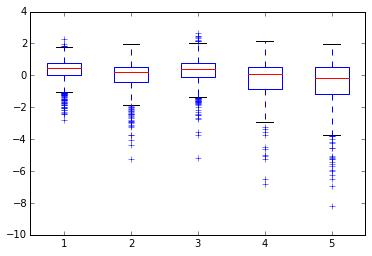
# ボックスプロット(最初の5サンプル)
_val = pylab.boxplot(ndata.values[:,0:5])
# 散布図(0番目と1番目、標準化前)
pylab.plot(data.iloc[:,0], data.iloc[:,1], 'go')
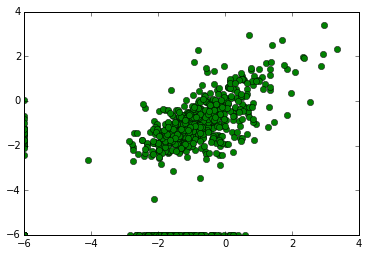
3つ目の引き数goは、greenで○の意味。詳しくは、matplotlibのドキュメントを参照してください。
データ解析¶
統計処理、PCA、K-meansクラスタリングなどをやってみます。
がんと正常組織で発現差のある遺伝子を見つけるために、まずはサンプルを分類して、別々のリストを作る。
tumor = []
normal = []
for v in ndata.columns:
if v[1] == 'T':
tumor.append(v)
elif v[1] == 'N':
normal.append(v)
else:
print(v)
何も出力されなければ、うまく分類できた。
# t検定
import scipy.stats as ss
ss.ttest_ind(data[tumor].ix['SYT3'], data[normal].ix['SYT3'])
(-2.0378779730286061, 0.042955723848096199)
出力はタプル。t統計量、P値の順。
主成分分析¶
# Scikit-learnを使ったPCA
from sklearn.decomposition import PCA
# 高次元データを、2次元に縮約
pca = PCA(n_components=2)
# 行方向にサンプルが並ぶように転置
pca_data = pca.fit(ndata.T).transform(ndata.T)
# 結果のプロット
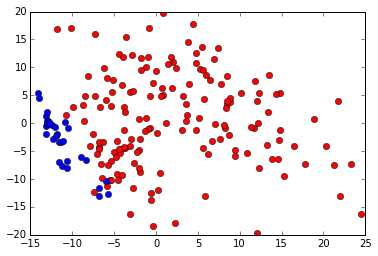
pylab.plot(pca_data[:,0],pca_data[:,1],'go')
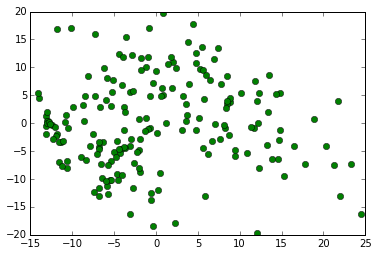
# がんのサンプルを赤く、正常組織を青く
for i,v in enumerate(ndata.columns):
if v[1] == 'T':
pylab.plot(pca_data[i][0], pca_data[i][1],'ro')
else: # Normal
pylab.plot(pca_data[i][0], pca_data[i][1],'bo')
K-meansクラスタリング¶
# K-meansクラスタリング
from sklearn.cluster import KMeans
# 先ほどのPCAと似たコードで実行できる。2クラスに分類
kmeans = KMeans(init='k-means++',n_clusters=2)
pred = kmeans.fit_predict(ndata.T)
print(pred)
[1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 0 1 1 1 1 1 1 0 1 1 0 1 1 1 1 0 0
1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 0 1 1 1 0 1
1 0 0 0 1 1 1 1 0 1 1 1 0 1 1 1 1 0 1 1 1 1 1 1 0 0 1 1 1 1 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0
0 0 0 0 1 0 0 0 0 1 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0]
# がんと正常がちゃんと分類できているか?(ちょっと高度な書き方)
from collections import Counter
Counter(zip(pred, [v[1] for v in ndata.columns]))
Counter({(1, 'T'): 89, (0, 'T'): 71, (0, 'N'): 31})
クラスタリングで1のクラスに入ったサンプルは、全部がんですが、0のクラスにサンプルが混在。2クラスにわけるのは失敗です。
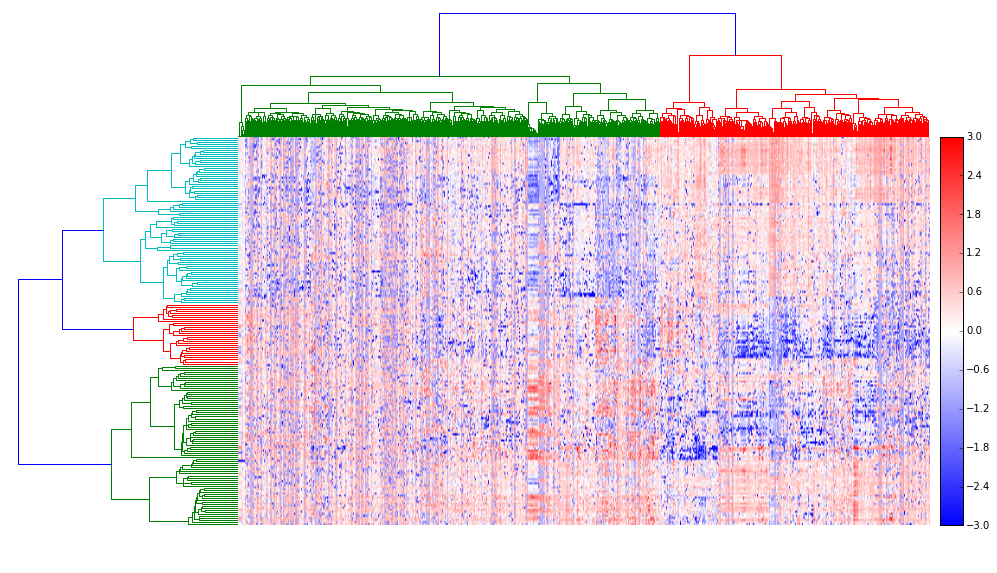
階層的クラスタリング¶
階層的クラスタリングを実行できるモジュールはありますが、樹形図とヒートマップを組み合わせる部分は自作が必要です。このコードはちょっと作りかけな感じがあるので、再利用するときは、ご注意ください。(近日中に直したいと思ってはいますが・・・)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 | import scipy
import scipy.cluster.hierarchy as sch
import scipy.spatial.distance as ssd
import matplotlib.cm as cm
import matplotlib.pyplot as plt
import matplotlib.colors as colors
def hc(data,file_name,both=True,index=False,index_names=None,annot=None,**kwargs):
fig = pylab.figure(figsize=(16,9))
if both:
ax1 = fig.add_axes([0.1,0.1,0.2,0.6])
Y = sch.linkage(data, metric='euclidean',method='ward') #sch.linkage(D, method='ward')
Z1 = sch.dendrogram(Y, orientation='right')
ax1.set_axis_off()
# Compute and plot sample dendrogram.
ax2 = fig.add_axes([0.3,0.7,0.6,0.2])
Y = sch.linkage(data.T,metric='euclidean' ,method='ward') #sch.linkage(D, method='ward')
Z2 = sch.dendrogram(Y)
ax2.set_axis_off()
# Plot distance matrix.
axmatrix = fig.add_axes([0.3,0.1,0.6,0.6])
if both:
idx1 = Z1['leaves']
idx2 = Z2['leaves']
D = data.values[idx1,:][:,idx2]
else:
idx2 = Z2['leaves']
D = data.values[:][:,idx2]
if file_name is None:
pylab.clf()
return D
im = axmatrix.imshow(D, aspect='auto', cmap=pylab.cm.bwr,vmin=-3,vmax=3)
#vmin=np.percentile(D.flatten(),5),
#vmax=np.percentile(D.flatten(),100))#cmap=pylab.cm.YlGnBu)
axmatrix.set_xticks(range(len(data.columns)))
axmatrix.set_yticks([])
axmatrix.set_axis_off() #全部消えてしまいます。
#print(data.columns.values[idx2,])
# hlines
# show sample name
col = [v[1] for v in data.columns.values[idx2,].tolist()]
axmatrix.set_xticklabels(col,rotation=90,size='xx-small',position=(0.0,-0.08))
#col = data.columns.values[idx2,].tolist()
#axmatrix.set_xticklabels(col,rotation=90,size='x-small',position=(0.0,-0.2))
# show row name
if index_names:
axmatrix.set_yticks([-0.5 + v for v in range(len(data)+1)])
yl = []
for v in data.index:
a = annot.ix(v[2])
_i = []
for k in index_names:
_i.append(str(a[k]))
yl.append('_'.join(_i))
axmatrix.set_yticklabels(yl,size=8)
elif index:
axmatrix.set_yticks([-0.5 + v for v in range(len(data)+1)])
axmatrix.set_yticklabels( ['chr%s:%d' % (str(v[0]),v[1]) for v in data.index],size='x-small')#,position=(0.0,-0.08))
# Plot colorbar.
axcolor = fig.add_axes([0.91,0.1,0.02,0.6])
pylab.colorbar(im, cax=axcolor)
fig.show()
fig.savefig(file_name)
#pylab.close(fig)
pylab.show()
return D
|
_d = hc(ndata.T,'temp.png')
まとめ¶
PyDataの紹介として、データの読み込みから、簡単なデータ解析までを概観しました。ちょっと突貫なので、暇を見つけてリニューアルします(たぶん) お気軽にお問い合わせください